Han transcurrido ya tres décadas desde el descubrimiento del SOD1 como uno de los genes causantes de la esclerosis lateral amiotrófica (ELA) de tipo familiar y, si bien hoy son ya más de 30 los genes identificados en la variante menos frecuente de ELA, todavía son muchos los aspectos de esta cruel y devastadora enfermedad que se siguen desconociendo ¿Qué causa la ELA? ¿Cuál es el desencadenante que provoca la muerte de las neuronas? Muchos los interrogantes alrededor de una enfermedad que, según la Sociedad Española de Neurología, afecta a entre 4.000 y 4.500 personas en nuestro país.
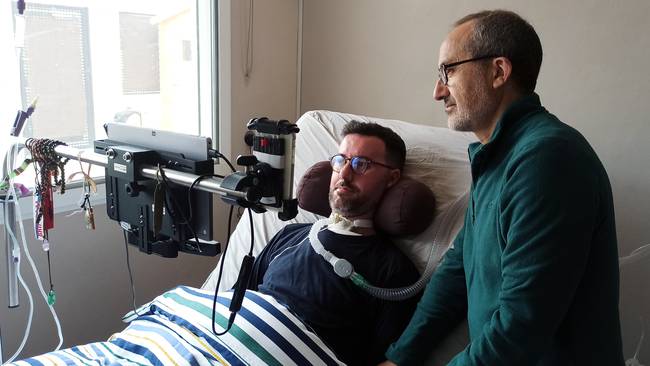
La ELA es una enfermedad neurodegenerativa irreversible y letal que se caracteriza por la degeneración progresiva y muerte de las neuronas motoras. Como consecuencia, poco a poco va paralizando prácticamente todos los músculos del cuerpo de quien la padece, de forma que la persona va dejando de andar, moverse, hablar, comer… hasta incluso de respirar sin ayuda. Frente a ello, su capacidad intelectual permanece intacta, por lo que el paciente es plenamente consciente de su deterioro.
El origen de la ELA se supone multifactorial, pero no tiene en la actualidad una causa conocida. Existen dos variantes: esporádica, la más frecuente (90% de los casos) y cuya aparición parece completamente azarosa y sin relación aparente con ningún tipo de factor de riesgo ambiental, profesional, geográfico, alimentario o cultural; y familiar, variante hereditaria que constituye entre el 5% y el 10% de los casos.
La ELA no tiene cura. De hecho, aunque se han probado multitud de fármacos de diferente tipo, ninguno ha logrado resultados positivos en los ensayos, a excepción del riluzol. Este medicamento, el único indicado en Europa para la ELA, fue aprobado por la Food and Drug Administration (FDA) norteamericana en diciembre de 1995 y, con resultados muy modestos, teóricamente aumenta la esperanza de vida entre tres y seis meses.
Existe también una posibilidad terapéutica aprobada en Estados Unidos y recientemente en Europa para pacientes con mutaciones en el gen SOD1 (presente en el 12% de los casos de ELA familiar y en el 1% de los de ELA esporádica) basada en administrar moléculas de ARN y que en ensayo clínico fase 3 parece ralentizar la progresión de la enfermedad.
En este escenario, la investigación y el avance en el conocimiento de la enfermedad son fundamentales para mejorar la calidad de vida de las personas que padecen ELA y para alcanzar la ansiada cura, investigación cuyos retos pasan por dar respuesta a los desafíos que se plantean a partir de la información que se desprende de los más recientes hallazgos relacionados con la identificación de más genes, el uso de biomarcadores y el incipiente apoyo de la epigenética para poder abordar la enfermedad en fases presintomáticas, sin olvidar nunca la situación de quienes padecen actualmente la enfermedad.
En línea con ello, decenas de grupos de investigación en todo el mundo están buscando en la actualidad marcadores sanguíneos y realizando comparativas genéticas para tratar de hallar algún parámetro que permita comprender si se va a producir la enfermedad o su progresión. Tras años con un único medicamento disponible, nos encontramos en un momento de gran movimiento en la investigación con muchos ensayos clínicos en marcha y en el que se están describiendo nuevas dianas terapéuticas. Encontrar la cura no está en un horizonte cercano, pero todas estas investigaciones nos permiten pensar en un próximo escenario con cada vez más tratamientos dirigidos a ralentizar la enfermedad.